

近日,人工智能学院崔振教授团队在《自然-机器智能》(Nature Machine Intelligence)上在线发表研究论文“Two-Dimensional Geometric Template Diffusion for Boosting Single-Sequence Protein Structure Prediction”。论文首次将三维蛋白质结构预测问题建模为二维视觉扩散学习任务,提出二维几何模板扩散框架TDFold,构建“二维图像到三维结构”重建的新范式,形成“蛋白质几何空间—视觉图像编码、二维拓扑几何—视觉扩散生成、三维结构—图网络解码”的系统化技术路径,实现了预测性能更优、资源消耗更低、推理效率更高的目标,为三维构象解析提供了全新解决方案。

人工智能技术的快速发展正加速驱动科学发现。三维结构智能解码技术正突破传统方法对海量同源信息的依赖,以更高效、更普适的方式揭示蛋白质结构规律,为精准医学、药物合成与研发等领域开辟新可能。传统主流AI方法(如同源建模)高度依赖已知同源信息,需通过大规模数据库搜索获取多序列比对和结构模板,不仅消耗大量计算资源与时间,成本高昂,更对缺乏同源数据的孤儿蛋白、快速进化的病毒蛋白形成了显著的技术瓶颈。
为解决上述问题,研究团队将视觉生成大模型引入蛋白质结构预测,提出二维几何模板扩散学习框架TDFold,形成了系统化的技术路径:
第一阶段:蛋白质几何空间到视觉空间的编码映射
利用二维矩阵与图像结构同构的特点,将二维几何模板矩阵信息离散化编码为图像的多通道特征表示,为引入预训练的视觉扩散模型(如Stable Diffusion)提供统一的表示基础。
第二阶段:序列引导的视觉扩散几何模板生成
受视觉生成模型启发,将蛋白质序列视为“文本描述”,二维几何模板视为“多通道图像”,借助LoRA适配器将蛋白质领域知识融入视觉扩散模型,使模型学会根据序列提示“绘制”几何图像,实现从序列到几何模板的生成式建模。
第三阶段:模板注入的双分支图网络三维重建
在获得二维几何模板作为结构约束后,采用双分支协同学习架构:一个分支从生成的几何模板中提取残基级空间约束,另一分支学习细粒度原子级特征。两路特征深度融合后送入SE(3)-等变图神经网络(SE(3)-EGNN),最终输出蛋白质三维结构(图1)。
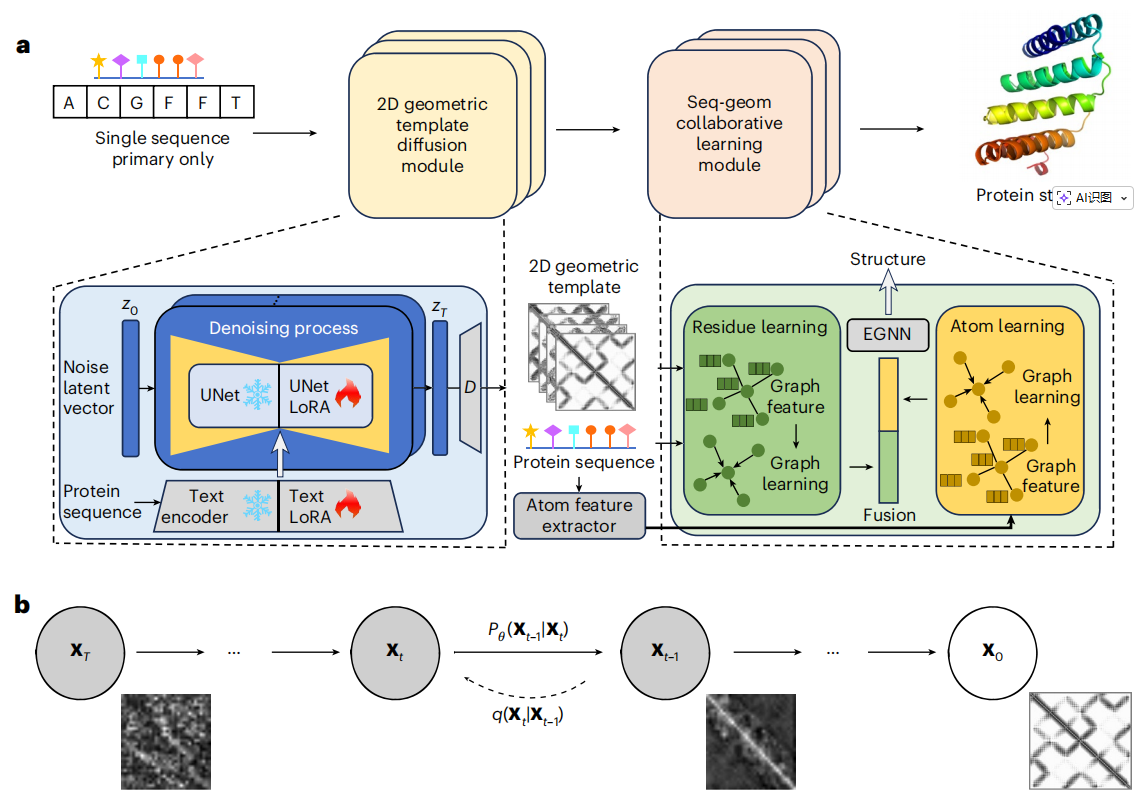
图1:TDFold模型架构以及二维模板扩散过程
在针对低同源性孤儿蛋白的预测任务中,TDFold在Orphan与Orphan25两个数据集上均显著优于现有对比方法。以2DCO_A和6XN9_A为例,传统方法检索到的模板与天然结构差异显著,而TDFold所生成的残基间距离矩阵与天然结构高度吻合,充分说明孤儿蛋白的生成几何模板质量远超搜索模板(图 2)。在同源性丰富的通用蛋白预测任务中,TDFold同样展现出卓越性能。在国际公认的CASP数据集上,采用单序列预测模式的TDFold取得了最优结果。以T1046s2-D1与T1106s2-D1为例,其生成的残基间距离图像与天然结构及搜索模板均保持高度一致,进一步验证了模型在通用蛋白场景下生成可靠几何信息的能力(图3)。上述结果表明,TDFold所构建的视觉生成模型能够有效学习蛋白质二维几何模板的数据分布,并依据氨基酸序列提示完成扩散生成,充分验证了将视觉扩散模型迁移至该技术范式的有效性与潜力。
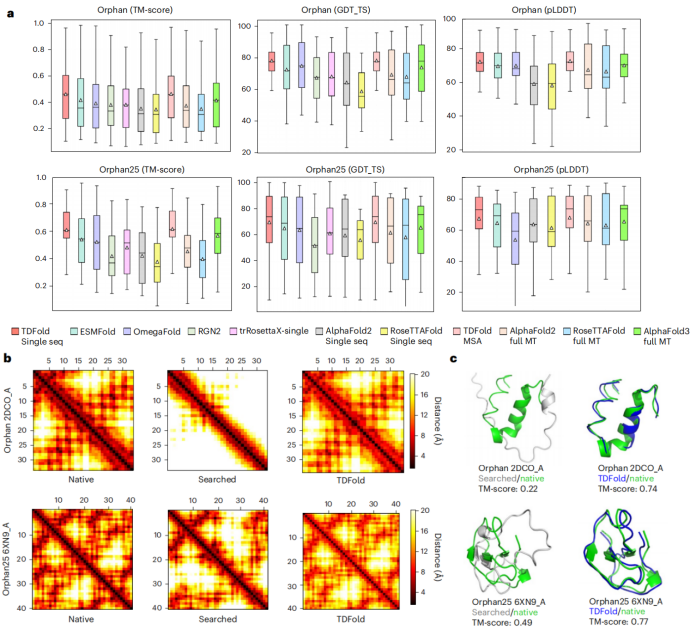
图2:TDFold在孤儿蛋白的二维几何模板生成和三维结构预测性能达到最优水平
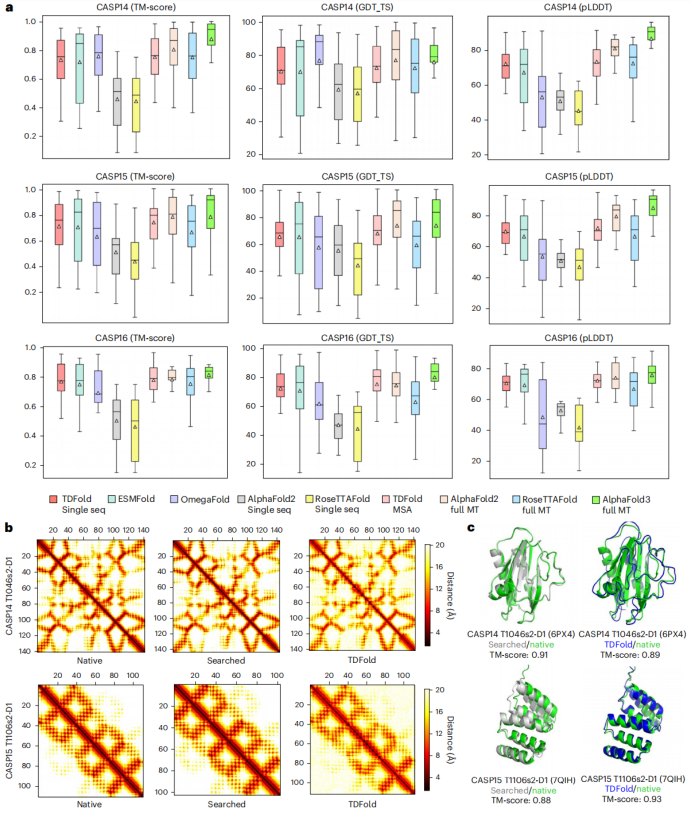
图3:TDFold在通用蛋白的二维几何模板生成和三维结构的单序列预测性能达到最优水平
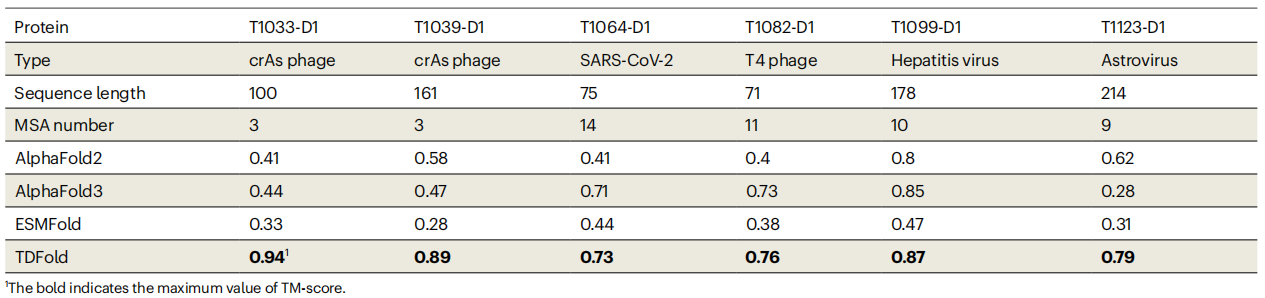
许多快速演化的病毒蛋白(如冠状病毒的非结构蛋白及辅助蛋白)毒株间序列同源性常低于20%,以逃避免疫应答并实现宿主适应性进化。这类蛋白在病毒致病中至关重要,但极低同源性给结构预测带来巨大挑战。本研究在CASP14–16病毒靶标(如SARS-CoV-2 ORF8辅助蛋白T1064,同源序列不足20)上测试发现,TDFold显著优于AlphaFold2、AlphaFold3及ESMFold,展现了同源信息匮乏下病毒结构预测的优越能力,对理解免疫逃逸机制及药物研发具有重要价值(表1)。
表1:TDFold在快速进化的病毒蛋白的三维结构预测均优于对比方法
在计算效率方面,TDFold展现出显著优势。以长度为500的蛋白质为例,TDFold推理仅需约10秒,相比耗时超1000秒的AlphaFold2/RoseTTAFold实现100倍加速,相比ESMFold(约100秒)也有10倍提升。显存占用方面,TDFold仅需约7GB,约为ESMFold(20GB)的30%。更关键的是,TDFold的资源消耗几乎不随序列长度增加而增长(长度从100增至500时,时间增幅<10%,显存增幅仅40%),而对比方法通常呈o(n³)增长。这些特性充分证明了tdfold在计算效率上的卓越优势(图4)。
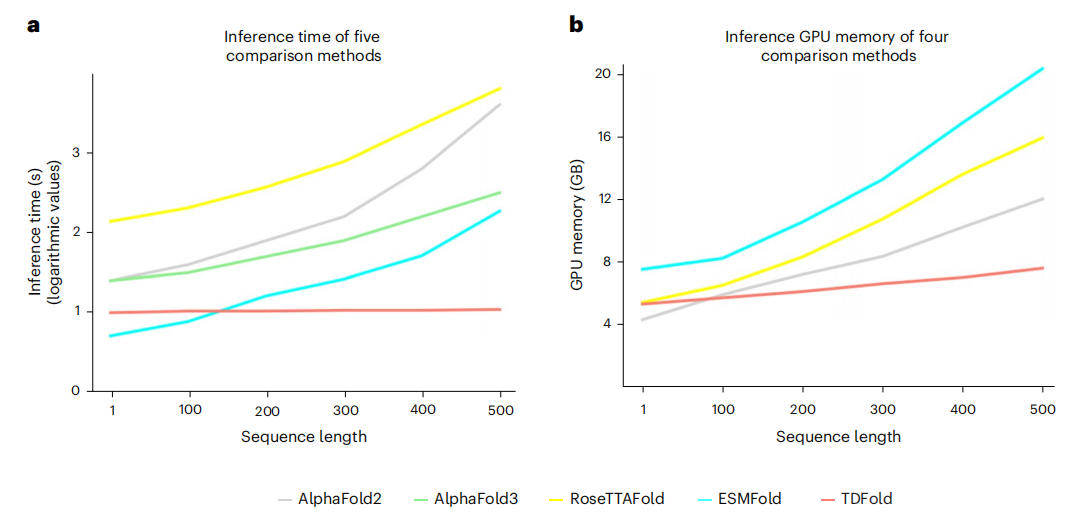
图4:TDFold在预测长序列蛋白质(=500氨基酸)的推理效率远优于对比方法
综上,本研究提出了一种基于二维几何模板扩散的蛋白质结构预测框架——TDFold。该框架能够从蛋白质序列提示中高效生成可靠的残基间几何信息,并利用序列与所生成的几何特征进行结构预测,从而省去了耗时的同源搜索步骤。TDFold对同源信息匮乏的序列(如孤儿蛋白、快速演化的病毒蛋白等)表现出较强的鲁棒性。该方法可在配备单张NVIDIA 4090(24GB)显卡计算机上完成高效训练与推理。其推理时间上的显著优势,使其尤其适用于高吞吐量的大规模预测任务。
北京师范大学人工智能学院教授崔振是独立通讯作者,南京理工大学计算机学院博士生王旭东和副教授张桐是该论文的共同第一作者。该工作得到了国家自然科学基金和中央高校基金的支持。